



 Download Factsheet
Download Factsheet Share
Share Contact
ContactPoint Source Carbon Capture

Advancing technologies optimized for cost, performance and reliability for the capture of CO2 from point sources, such as fossil fuel-based power generation and industrial facilities
The U.S. Department of Energy/National Energy Technology Laboratory’s (DOE/NETL) Point Source Carbon Capture (PSCC) Program is developing the next generation of advanced carbon dioxide (CO2) capture technologies (e.g., membrane, solvent, sorbent, chemical looping) focusing on reduced cost and improved performance and reliability. DOE’s Office of Fossil Energy and Carbon Management (FECM) has adopted a comprehensive, multipronged approach that involves the coupling of CO2 capture from a point source (fossil fuel-based power generation and industrial sources, e.g. hydrogen, petrochemical, cement production plants) with (1) CO2 transport via pipeline and injection underground for long-duration storage, (2) conversion into valuable products such as fuels, chemicals and building materials, or (3) use for enhanced hydrocarbon (oil or gas) recovery.




The PSCC Program is investing in innovations to drive down costs and optimize processes to enable widespread use of these technologies to generate CO2 that can support the expansion of secure domestic energy production. Research and development (R&D) efforts to date have led to significant improvements in cost and performance through implementation of energy and process efficiencies and development of advanced CO2 capture media (e.g., solvents, sorbents, and membranes). The program focuses on developing highly efficient, scalable carbon capture technologies with even greater cost reductions, that are capable of flexible and reliable operation.
Point Source Carbon Capture

Capture from Power Generation Sources
PSCC in fossil fuel-based power production separates CO2 from the plant’s exhaust gas or syngas stream. The PSCC Program focuses on advancing novel carbon capture materials, equipment, processes or a combination thereof for applications in natural gas combined cycle and coal-based power generation. R&D efforts involve development and testing of highly efficient component designs, material systems and/or integrated processes.

Capture from Industrial Sources
PSCC from industrial facilities, such as cement manufacturing, hydrogen production and petrochemical plants, with minimal impact on the cost of product, is a vital element in enhancing the competitiveness of American industry. R&D efforts are focused on the development and testing of transformational technologies for industrial carbon capture.
Key Analysis Areas
Solvents
Solvent-based CO2 capture involves chemical or physical absorption of CO2 from a gas into a liquid carrier. Development of advanced solvents (e.g., water-lean solvents, phase-change solvents, high-performance functionalized solvents) that have a lower regeneration energy requirement than existing amine systems, combined with high CO2 absorption capacity and tolerance to impurities, is a key objective for lowering capture costs. System advancements include process intensification techniques, methods to mitigate aerosol formation and corrosion, and heat integration approaches.

Sorbents
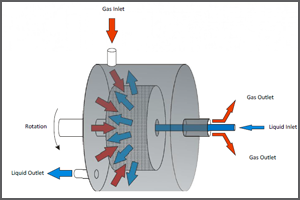
Sorbent-based CO2 capture involves the chemical or physical adsorption of CO2 from a gas using a solid sorbent. R&D objectives include development of low-cost durable sorbents that have high selectivity for CO2, high CO2 adsorption capacity, resistance to oxidation, and can withstand multiple regeneration cycles with minimal attrition. System advancements include sorbent process intensification techniques, novel reactor designs, and enhanced process configurations, such as rotating beds for CO2 adsorption and desorption.

Membranes
Membrane-based CO2 capture uses permeable or semipermeable materials that allow for the selective transport and separation of CO2 from a gas. Membrane processes offer potential advantages when compared to other CO2 separation technologies, including no hazardous chemical storage, handling, disposal, or emissions issues; simple passive operation; tolerance to high sulfur oxide and nitrogen oxide content; a reduced plant footprint; and efficient partial CO2 capture. R&D objectives include development of low-cost, durable membranes (e.g., polymeric membranes, mixed matrix membranes, sub-ambient temperature membranes) that have improved permeability and selectivity for CO2, thermal and physical stability, and tolerance to gas contaminants. Process enhancements for membrane-based capture systems include low-pressure drop membrane modules.

Novel Concepts
Novel concepts include alternative technologies and processes, such as cryogenic separation and electrochemical membranes, and additive manufacturing of novel system components and materials. R&D objectives include development of equipment, materials, and processes that enable intensified thermodynamic operations, improve process performance, and reduce equipment size, lowering capital and operating costs.

Hybrid
Hybrid systems efficiently combine two key technologies in a single system (e.g., sorbent-membrane system). Hybrid concepts can reduce the process’ overall energy intake by leveraging process synergies, resulting in a more cost-effective system.
Enabling Technologies
Enabling technologies are concepts that could improve a whole class of materials, and although the research might be applied to one specific material, it is envisioned that substantial research findings could benefit multiple materials. R&D topics include solvent aerosol emissions mitigation, solvent viscosity reduction, solvent stability improvements, materials compatibility, corrosion resistance improvements, and reduction or separation of capture media degradation products.






